
David Kilper/WUSTL
The Washington University in St. Louis team participating in the modENCODE project, a massive ongoing effort to map all the elements in living cells that affect whether genes are expressed or silenced, discuss a fruit fly strain with an enzyme that affects gene expression. Team members are (from left): Sarah C.R. Elgin, PhD, the Viktor Hamburger Distinguished Professor in Arts & Sciences who led the team; Sarah Gadel, lab technician; Nicole C. Riddle, PhD, research assistant professor of biology in Arts & Sciences; and Tingting Gu, PhD, a postdoctoral research associate in biology. Missing is Sarah Marchetti, a technician. A major paper that includes their part of the modENCODE project has just been published in Nature.
In 2003, the year a complete draft of the human genome was released, the U.S. National Human Genome Research Institute launched the ENCODE project (ENCyclopedia of DNA Elements), to develop an encyclopedia of the epigenome, that is, of all of the many factors that can change the expression of the genes without changing the genes.
Four years later, the National Institutes of Health funded modENCODE (the Model Organism ENCylopedia of DNA Elements) to work out the epigenomes of two model organisms: the fruit fly Drosophila melanogaster, lurker among rotten bananas, and the round worm, Caenorhabditis elegans, slitherer between crumbs of soil.
This was big science with a vengeance. The work had been divided among 21 teams, themselves each made up of multiple labs, all toiling away at universities and companies across the country. And that’s not counting the research groups tasked with coming up with new ways of handling the floods of data pouring out of the labs.
What was going on?
Sarah C.R. Elgin, PhD, the Viktor Hamburger Distinguished Professor in Arts & Sciences, who led the Washington University in St. Louis lab that is part of one of the modENCODE teams, offers an explanation.
“We learned many things from the Human Genome Project,” Elgin says, “but of course it didn’t answer every question we had.
“Including one of the oldest: We all start life as a single cell. That cell divides into many cells, each of which carries the same DNA. So why are we poor, bare, forked creatures, as Shakespeare put it, instead of ever-expanding balls of identical cells?
“This work,” says Elgin, “will help us learn the answer to this question and to many others. It will help us to put meat on the bones of the DNA sequences.”
What is the epigenome?
Instead of spewing out long strings of the As, Ts, Gs and Cs like the gene sequencing labs, the epigenetic labs are disgorging voluminous data about the proteins bound to the DNA and the many other gizmos and widgets that make up the machinery of gene expression.
These widgets are collectively called the epigenome, because they provide a level of control in addition to, or beyond, the level provided by the genome. Whereas the genome is the same in every cell of an organism, the epigenome of every cell type is different. It is because of the epigenome that a liver cell is not a brain cell is not a bone cell.
By 2007, ENCODE had analyzed the epigenetics of about one percent of the human genome and published the results in Nature and a special issue of Genome Research.
To speed things up, the National Institutes of Health funded modENCODE to work out the epigenetics of the fruit fly and the round worm C. elegans.
Both organisms, which have much smaller genomes, will provide reference points for the more intimidating project of unraveling the much larger human epigenetic system. They also provide a quick and relatively easy way of verifying hypotheses arising out of the ENCODE data.
The first results from the modENCODE projects, which were divided among 11 teams, have just been published in Science and Nature. Many more papers are forthcoming.
Led by Gary Karpen of the Lawrence Berkeley Lab and the University of California at Berkeley, the modENCODE chromatin group includes teams led by WUSTL’s Elgin, Mitzi Kuroda of Harvard Medical School, Peter Park of Harvard Medical School and Vince Pirrotta of Rutgers University. Elgin’s team is made up of Nicole C. Riddle, PhD, research assistant professor of biology in Arts & Sciences; Tingting Gu, PhD, a postdoctoral research associate in biology; and Sarah Gadel and Sarah Marchetti, lab technicians.
Karpen’s team has just published a comprehensive description of the chromatin landscape of the fruit fly genome in Nature, which provides a representative example of the science coming from the larger undertaking.
The chromatin landscape
So why are we poor, forked creatures instead of tumbling beachballs of undifferentiated cells?
An embryo begins by dividing into identical cells, but within hours these cells begin to make genetic decisions, turning off some genes and turning on others. So the ball of cells acquires a front end and a back end, a top and a bottom, nerve cells and muscles cells — all still carrying the same DNA, but DNA now packaged in such a way that some genes are shrink wrapped and silent but others are spreadeagled for easy access and active.
We’ve been taught that DNA is everything, but you could equally well say packaging is everything.
And in eukaryotes (higher organisms like plants and animals), DNA comes in a particularly elaborate package.
In human beings, nucleus of a cell, which is only a few micrometers in diameter, contains two meters of DNA! Trying to get in all that DNA in the nucleus is like packing a thread 10 kilometers long into a pea.
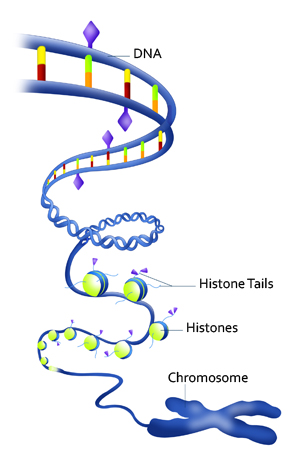
To keep things manageable, the DNA is wound around tiny beads of proteins called histones and these “beads on a string” are then coiled and folded, looped, and then coiled and folded again to “condense” the DNA into a chromosome.
The combination of DNA, histones and other proteins that make up chromosomes is called “chromatin.”
The epigenetic code that determines whether genes are silenced or expressed consists of chemical modifications to the DNA, to “tails” that hang off the histones, or other packaging proteins.
“ENCODE and modENCODE are much more complicated projects than the Human Genome Project,” Elgin says, “because the DNA sequence is pretty much the same in every cell type, whereas the chromatin structure is different in every cell type. In fact we believe it is the chromatin structure that differentiates one cell type from another.
“That means we can’t just do one genome for the organism. We have to do every different cell type to get a complete picture of the organism, and that’s a daunting prospect.”
ChIP experiments
“In the Nature experiment,” Elgin continues, “we started by looking at two different types of fruit fly cells, called S2 and BG3, that we could grow in tissue culture. By culturing the cells, we ensured we would have an abundant supply of cells whose chromatin structure we could be reasonably certain was always the same.”
The cells were then run through a highly automated process to map the epigenetic marks on the DNA. For this study, the tool of choice was the chromatin immunoprecipitation or chIP array.
“In a typical experiment,” Elgin says, “we expose the chromatin to formaldehyde, which binds the packaging proteins to the DNA. Then we shear the DNA into little fragments.
“And then — this is the key step — we put in an antibody to a particular protein modification, a chemical that binds to that protein.
“You have to be sure that your antibodies are good,” Elgin says. “We went to a lot of trouble on that issue. The antibodies must recognize one protein and only that protein. That’s absolutely crucial.
“Now we use that antibody to fish out all the fragments that have that modified protein and we put them in one pot, discarding all the rest of the fragments. Next we purify the selected DNA, throwing away the proteins, and we identify the fragments of selected DNA by putting them on a microarray.” (see illustration)
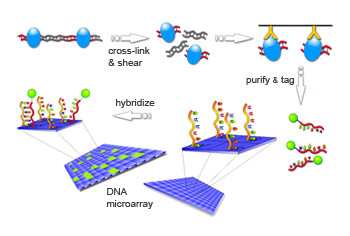
Wikimedia Commons
In a chIP experiment, antibodies to proteins bearing particular epigenetic marks are used to fish them out of the cell. The chemical formaldehyde binds proteins (blue pods) to neighboring DNA (top left). The cells are then broken open and the DNA is cut into fragments. Antibodies (yellow Ys) specific to the proteins then grab the protein-DNA complexes and affix them to a solid surface (top right). The proteins are discarded and the double-stranded DNA molecule is unzipped and each half is tagged with a fluorescent molecule (green ball). The tagged DNA fragments are then poured over a DNA microarray which holds single-stranded fragments of fruit fly DNA. Whenever a labeled fragment finds a matching fragment on the array, the two fragments zip together. The labeled fragments glow under fluorescent light (bottom left). The light signal is captured by a camera, and converted to numerical values that can be analyzed by a computer.
The microarrays are read at Vincenzo Pirrotta’s lab at Rutgers, and the data are sent to Peter Parks’ lab at the Center for Biomedical Informatics at Harvard Medical School where computers map the marks onto the known fruit fly genome.
“You could not do this kind of biology without computers,” Elgin says. “No way. The computer is an essential tool if you’re going to be a biologist. Times have changed!
“We then do the same experiment over again but with different antibodies. And we keep doing the experiment again and again and sending the data to the bioinformatics people.”
As the data build up, the computer begins to look for groups of modifications that tend to occur together. For example, different sets of modifications might occur on genes that are repressed, paused, fluctuating or active.
In the end, the team found they were able to capture most of the complexity of the fruit fly’s chromatin landscape with nine combinatorial states, that is, nine sets of DNA or protein modifications.
Beautiful chromosomes
Drosophila has a pretty simple genome, Elgin says, consisting of only four chromosomes.
The chromatin states can be visualized by drawing long, thin chromosomes and coloring them in bars that correspond to different chromatin states. But these vermicelli-like graphs are hard to read.
Another way to visualize the states is to fold the chromosomes into a geometric pattern that maintains the spatial proximity of nearby regions of the chromosome. This makes it easier to spot patches of uniform color that correspond to domains with the same chromatin state.
In these maps of the fruit fly’s chromatin, differences are immediately evident.

The fruit fly has four pairs of chromosomes: an X/Y pair (the chromosomes in the illustration are from a male fly cell but just the X is shown because the Y is very difficult to map) and three autosomes labeled 2, 3, and 4. The fourth chromosome (bottom right) is so tiny that it is often ignored.
The dark blue in the lower right corner of the left arm of the fruit fly’s third chromosome (left middle) corresponds to heterochromatin, a tightly packed, usually silent form of DNA that forms around the structure called the centromere where the two arms of a chromosome are joined. (The centromere is the site where the chromosomes attach to the mitotic spindle during cell division.)
“The flecks of red mark transcription start sites,” Elgin says, “spots where we find genes that are being actively expressed. They are usually flanked by states 2, 3 and 4 which we think are related to gene regulation. They include the enhancers, regions of DNA that facilitate the transcription of genes.
“The X chromosome (bottom left) is clearly special,” says Elgin. “It’s got a lot of the green state, which we think is part of dosage compensation. Sex is determined differently in flies than in people. If a fly has one X chromosome, it is male. If it has two it is female. The Y chromosome exists but is not involved in sex determination.
“Because male and female flies have different numbers of copies of the X chromosome genes,” Elgin says, “something must be done to adjust gene expression based on the fly’s sex. That’s what we think the special mark for X is all about.”
Elgin is particularly interested in chromosome 4 (bottom right). This tiny chromosome is almost entirely silenced (the dark blue state), and yet, she says, there are red marks (active transcription start sites) sprinkled throughout the sea of blue. “These are 80 active genes, genes that must have some kind of mechanism for elbowing aside silencing marks at the start of transcription,” she says. “We don’t know how that happens, but it’s one of the things we’d really like to get at. “
In bacteria, Elgin says, most of the DNA consists of genes; it actually codes for proteins. But in our genome, only a small percent of the DNA actually codes for protein. Most of it — as much as 95 percent — is silenced to some degree, expressed at a low level if at all. Much of this DNA appears to be derived from retroviruses, devious viruses able to convert their RNA to DNA and insert it in our genome, or from DNA transposons, small bits of DNA that can move around in the genome.
“We know that silencing this DNA is essential for maintaining the stability of the genome,” Elgin says. When bits of it lose their packaging and move about they can cause devastating diseases, such as some forms of muscular dystrophy. Conversely if the silencing mechanism is tricked into making a mistake and shuts down an important gene, the results can be equally detrimental; this is apparently what happens in Fragile X syndrome.
Zen-like, she concludes that silence may be as important as expression. “It’s like sculpture — what you see depends not on what you add, but on what you take away.”