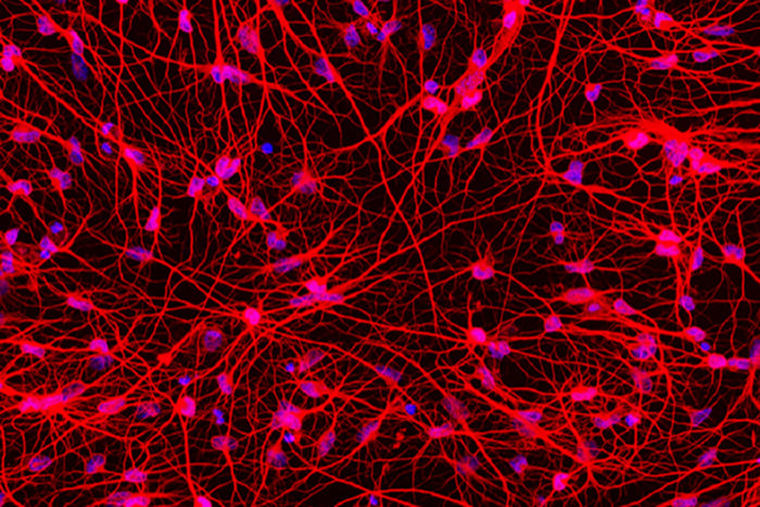
Scientists at Washington University School of Medicine in St. Louis have transformed skin cells from patients with Huntington’s disease into the type of brain cell affected by the disorder. The resulting mass of neurons serves as a new tool to study the degenerative and eventually fatal neurological condition, according to the researchers.
The study, published Feb. 5 in Nature Neuroscience, showed that the patients’ nerve cells — converted directly from patients’ skin cells — exhibited “symptoms” of the disorder, including DNA damage, dysfunctional mitochondria and cell death. Correcting for malfunctioning genes in these reprogrammed neurons prevented the cell death that is characteristic of Huntington’s disease, an inherited genetic disorder that causes cognitive decline and involuntary muscle movements. Symptoms typically appear in people with the disease when they are ages 30 to 50 and steadily worsen over time. On average, patients live about 20 years after symptoms begin.
“This is a powerful tool to investigate the reasons why particular brain cells with the disease-associated mutation become sick over time and eventually die,” said senior author Andrew S. Yoo, assistant professor of developmental biology. “In theory, we could model progression of the disease by reprogramming skin cells from patients at a range of ages, including before symptoms begin. And if there are drugs or compounds that may help these patients, we can test them first in this system.”
Huntington’s disease and other inherited brain conditions are challenging to study because it is difficult to obtain samples of neurons from living patients. Seeking the next best thing, scientists have found ways to transform skin cells into brain cells.
Skin cells are easy to collect from patients and share the same genetic blueprint — and disease-causing mutations — as brain cells. The researchers, including first author Matheus Victor, a postdoctoral research associate, set out to generate neurons that would mimic those of adult patients in order to model the onset and progression of Huntington’s disease. They achieved this goal with a direct conversion method they developed.
The method allows skin cells to bypass the stem cell stage as they are being reprogrammed into neurons. Passing through a stem cell stage resets the developmental clock to an embryonic-like state, wiping out the age-associated effects of the disorder. But the directly reprogrammed neurons retain their age, along with the problems associated with adult-onset Huntington’s disease, according to Yoo and his colleagues.
In the direct reprogramming, the researchers exposed the adult skin cells to a specific mix of signaling molecules the scientists’ past research had found would convert healthy skin cells directly into a type of brain cell called medium spiny neurons, without intermediate steps along the way. This signaling cocktail repackages the DNA, folding up the instructions for skin cells and unfurling the instructions for neurons. In this method, the resulting reprogrammed medium spiny neurons retain the patient’s chronological age, along with age-associated symptoms of the disorder.
Although other neuronal cell types are affected, medium spiny neurons bear the brunt of the damage caused by Huntington’s disease. The genetic error that causes the disease leads a key protein, Huntingtin, to be misshapen and not function properly. As a result, the malfunctioning protein builds up and, by some set of events that remains unknown, eventually kills the cell. Since the neuronal death can be recapitulated in directly reprogrammed patient neurons, Yoo said the new technique offers a way to study the details of how potential therapies — including drugs that are currently being tested in clinical trials — could rescue medium spiny neurons from death.
With a focus on understanding the details of how the disease progresses to cause cell death, Yoo and his colleagues identified another important protein. The protein, called SP9, was known to be important for normal medium spiny neurons, but it had not previously been associated with Huntington’s disease. The researchers found significantly less SP9 in the transformed medium spiny neurons from Huntington’s patients. They performed an experiment in which they gave back SP9 to the diseased cells and found that returning this protein to the diseased neurons reduced cell death to levels similar to healthy control cells.
“We want to understand what drives the disease progression over time,” Yoo said. “Most neurodegenerative disorders worsen over time, so this method of modeling potentially could be applied in other conditions. This technique lets us capture characteristics of the disease at distinct moments in its progression. That’s important in understanding what is happening and finding ways to stop it.”